 Cell Technology Co., Ltd.")

News
News Center
China's Practices in Developing Stem Cell Drugs in Compliance with FDA Regulatory Requirements
2020-03-16
You may have heard of the FDA in the news, or seen the words "FDA-approved" in other advertisements—so what exactly makes the FDA so remarkable? And why is "FDA certification" considered the highest honor and guarantee of product quality?

Today, the editor will take you on a quick tour.
What is the FDA?
FDA stands for the Food and Drug Administration, and it sometimes refers specifically to the U.S. Food and Drug Administration. Authorized by the U.S. Congress—acting as the federal government—the FDA is the nation’s premier regulatory agency responsible for overseeing the safety and efficacy of food and pharmaceutical products. It is also a government health-regulation body staffed by professionals such as doctors, lawyers, microbiologists, chemists, and statisticians, all dedicated to safeguarding, promoting, and enhancing public health. Many other countries rely on seeking and receiving FDA assistance to ensure and monitor the safety of their own domestic products.
How authoritative is the FDA, exactly?
The FDA is one of America's earliest consumer protection agencies, wielding immense influence both in the U.S. and around the world—it’s often referred to as the "Guardian of American Health." With its unwavering dedication to safeguarding and fulfilling its mission and responsibilities, the FDA has become an impenetrable shield in the hearts of food and drug consumers globally.
Since 1990, the U.S. FDA has closely collaborated with international organizations such as ISO, continuously driving a series of innovative measures. Particularly in the food and pharmaceutical sectors, FDA certification has become the world’s gold standard for food and drug safety testing—and is recognized by the World Health Organization as the highest global benchmark for food safety. Only products that undergo rigorous monitoring across 143 critical testing points after being used on humans, coupled with ongoing surveillance of 20,000 to 30,000 individuals over a period of 3 to 7 years, are granted FDA certification if they consistently meet all stringent requirements.
Therefore, many international manufacturers regard obtaining FDA approval as the highest honor and guarantee of product quality.
How are new drugs regulated by the FDA in the United States?
The FDA was established in 1906; prior to that, U.S. drugs were completely unregulated and marketed solely through advertising. In 1938, it became mandatory for drug manufacturers to prove their products' safety before they could be sold. Then, in 1962, the requirement was further strengthened: drugs now had to demonstrate not only safety but also efficacy before being approved for sale. The FDA has the authority to inspect manufacturing facilities and can prosecute those who violate regulations.
Through continuous development, the U.S. FDA has established the most detailed and comprehensive operational structure in the world, comprising 12 offices and bureaus, as well as 7 centers. While the 12 offices and bureaus will not be elaborated on here, let’s focus specifically on the Center for Drug Evaluation and Research (CDER), one of the seven centers—namely, the Center for Food Safety and Applied Nutrition, the Center for Veterinary Medicine, the National Toxicology Program, the Center for Biologics Evaluation and Research, the Center for Herbal Products, the Center for Drug Evaluation and Research, and the Center for Devices and Radiological Health.

The U.S. FDA has 7 centers under its umbrella.
CDER is one of the largest review centers within the U.S. FDA, dedicated to ensuring the safety and efficacy of both prescription and over-the-counter medications. It oversees most of the drugs defined under the Federal Food, Drug, and Cosmetic Act. The center is responsible for evaluating applications for both new and generic drug approvals, enforcing cGMP regulations to govern pharmaceutical manufacturing in the United States, and determining which medications require a doctor’s prescription. Additionally, CDER monitors the accuracy of drug advertisements broadcast on television, radio, and in print media, while also collecting and analyzing real-world safety data on marketed drugs. Through its rigorous oversight, CDER ensures that consumers receive accurate, reliable, and safe information about the medicines available to them.
The FDA's review mechanisms and efficiency are both worthy of our reference and learning—just in 2019 alone, the FDA approved 48 new drugs, maintaining the momentum of recent years.
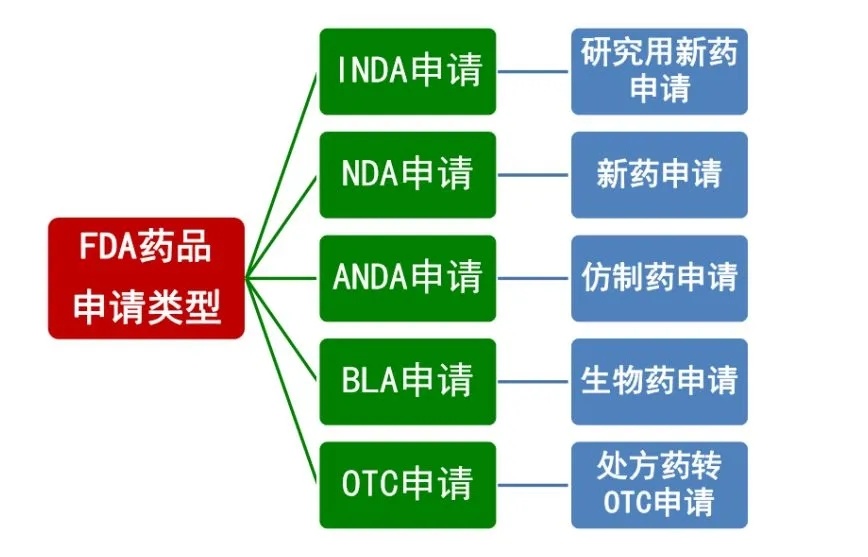
FDA's types of drug applications
Section 505(i) of the Food, Drug, and Cosmetic Act states that drugs developed specifically for research purposes by qualified experts are exempt from the requirements of an NDA or eligible for a streamlined New Drug Application (ANDA). Applying for this exemption is known as an Investigational New Drug (IND) application. For new IND applications, there is a mandatory 30-day safety review period—meaning the FDA has 30 days to evaluate the drug’s safety before proceeding.
IND application information must include (the most basic requirements for Phase 1):
1. Animal Pharmacology and Toxicology Studies (Safety Requirements)
• Ensuring safety in human testing
2. Production Information
• Ingredients, manufacturer, stability, control measures, etc.
• Ensuring the production and supply of consistently uniform batches of pharmaceuticals
3. Clinical Protocol and Investigator Information
The stages of IND:
Phase 1: First-time use of the new drug in humans. This phase primarily focuses on evaluating the safety of the stem-cell-based drug being tested in clinical trials, studying how the medication is metabolized and pharmacologically active in the human body, identifying potential adverse reactions associated with dose escalation, and gathering early evidence of efficacy. The process is closely monitored by the FDA, and typically involves 20 to 80 patients or healthy volunteers in the clinical trial.
Phase 2: Restricted and Controlled Clinical Studies. This phase primarily evaluates the preliminary effectiveness of the drug being tested in clinical trials, typically compared against a placebo. The goal is to gather early data on the drug’s efficacy and evidence of short-term safety, with no serious adverse effects reported. As with earlier phases, this stage remains under close FDA oversight, and the number of participants usually ranges in the hundreds.
Phase 3: Expanded, Controlled, and Uncontrolled Trials. This phase primarily focuses on evaluating the drug's precise efficacy, investigating its pharmacological effects, metabolism, pharmacokinetics, and identifying potential adverse reactions—both individually and in combination with other medications. It also involves comparing the new drug to existing, similar treatments already on the market, followed by a comprehensive benefit-risk assessment. The ultimate goal is to gather additional insights into the drug’s effectiveness and safety profile, enabling extrapolation of these findings to the broader population. These insights will then be translated into clear, evidence-based labeling information for healthcare professionals, providing a robust foundation for clinical decision-making. Typically, this phase enrolls anywhere from several hundred to several thousand participants.
Once all the above stages are completed, you can proceed with the new drug application. If approved and the on-site inspection is successfully passed, the drug will receive FDA approval for market release.
Jiuzhitang Maker's stem cells meet FDA requirements.
In February 2020, Jiuzhitang Maker’s clinical trial for a new stem-cell drug received approval—marking the first time the Center for Drug Evaluation under China’s National Medical Products Administration has approved a clinical trial using imported stem cells, the first to employ bone marrow-derived mesenchymal stem cells, and also the first to explore stem-cell therapy for a major neurological indication. This milestone holds significant importance for the development of China’s stem-cell industry.
The stem cell product used in this clinical trial is ischemia-tolerant allogeneic human bone marrow mesenchymal stem cells (ithMSCs), manufactured by Stemedica Cell Technologies, Inc. of the United States. Founded in 2005, Stemedica received a manufacturing license from the U.S. regulatory authority in 2010 and has been operating under GMP-compliant conditions for nearly 10 years. It is one of the few companies worldwide capable of producing both bone marrow mesenchymal stem cells and neural stem cell products under cGMP standards. Notably, the company’s stem cells have been recognized by Life Technologies (now part of Thermo Fisher Scientific) as "best-in-class" products.
Stemedica’s ithMSC product, produced under fully controlled hypoxic conditions, meets U.S. cGMP standards and FDA requirements for both its manufacturing process and quality system. Additionally, Stemedica’s bone marrow-derived mesenchymal stem cell products have already received 7 IND approvals in the U.S., meaning these 7 clinical trials are being conducted under FDA approval and oversight.
Stemedica and its partners are conducting 17 clinical trials across multiple countries, targeting a range of conditions including stroke, chronic heart failure, acute myocardial infarction, skin photoaging, type 2 diabetes, osteoarthritis, traumatic brain injury, and Alzheimer’s disease. Notably, their treatment using ischemia-tolerant allogeneic human bone marrow mesenchymal stem cells for acute myocardial infarction has completed Phase III clinical trials in Kazakhstan and has since received approval from the Kazakh Ministry of Health for commercialization.

Stemedica's global R&D pipeline for bone marrow mesenchymal stem cells and neural stem cell products Note: Stemedica has licensed its cardiac disease indication to partner CardioCell LLC. )
Jiuzhitang Maker (Beijing) Cell Technology Co., Ltd., as the sole Chinese partner of U.S.-based Stemedica Corporation for stem cell technology, has acquired the core technologies enabling scalable, standardized, and traceable stem cell production by introducing Stemedica’s globally advanced, clinical-grade stem cell preparation platform—thus filling a critical gap in China’s domestic market.
Meike has established a production platform at the Beijing Daxing Biomedical Base—known as China's "Pharmaceutical Valley"—that complies with both Chinese and U.S. cGMP-quality standards, enabling it to manufacture commercial-grade stem cells meeting drug-approval requirements in both China and the United States.
Next, Jiuzhitang Maker will collaborate with Beijing Tiantan Hospital, Affiliated to Capital Medical University, to conduct a clinical trial for the treatment of ischemic stroke.
Related News

Here is the title—h1 placeholder text


Copyright © Jiuzhitang Maker (Beijing) Cell Technology Co., Ltd.
Powered by: 300.cn SEO | Privacy Policy