 Cell Technology Co., Ltd.")

News
News Center
Big news! The National Medical Products Administration has released the "Drug Production Quality Management Regulations—Appendix on Cell Therapy Products (Draft for Comments)."
2022-01-10
On January 6, 2022, to implement the "Drug Administration Law of the People's Republic of China," the National Medical Products Administration (NMPA) publicly released the draft "Appendix to the Pharmaceutical Production Quality Management Regulations—Cell Therapy Products" for public comment through its Comprehensive Department, inviting input from society.
The draft for soliciting opinions clearly states that the cell therapy products referred to hereinafter as "cell products" are defined as: Human-derived live cell products, including cells that have or have not undergone genetic modification—such as autologous or allogeneic immune cells, stem cells, tissue cells, or cell lines, among other products. , excluding blood components used for transfusion, allogeneic hematopoietic stem cells already regulated for transplantation, reproductive-related cells, as well as tissue and organ products composed of cells, among others.
According to the draft for comments, the appendix applies to The entire process of cell product handling, from the transportation and receipt of donor materials, through product manufacturing and testing, to final release, storage, and shipment. The processes for the production, testing, and release of gene-modified vectors or other starting biological materials (including viruses, plasmids, RNA, antigen peptides, antigen proteins, protein-RNA complexes, and more) intended directly for cell product manufacturing must comply with the requirements outlined in the current version of the "Pharmaceutical Production Quality Management Regulations," its relevant appendices, and this supplementary appendix.
Notably, the Draft for Soliciting Opinions proposes Requirements for personnel, materials and products, as well as production management. According to these guidelines, the head of production management, the head of quality management, and the quality authorized person must possess relevant expertise (such as microbiology, biology, immunology, biochemistry, or biopharmaceutical sciences) and be fully capable of fulfilling their responsibilities in both production and quality management. Enterprises should conduct risk assessments for materials, and the identification of critical materials must be documented. Incoming inspections should be carried out for critical materials, with specific risks and additional risk-mitigation measures (such as enhanced quality control) taken into account. Furthermore, operational procedures must be established for the safe and efficient handling of non-compliant donor materials, intermediate products, and finished goods—and all handling activities should be properly recorded. If adjustments to cell product manufacturing process parameters are necessary due to inherent quality variations in donor materials, enterprises must ensure that such adjustments remain within the scope approved during product registration.
— The full text follows —
☟
Good Manufacturing Practice for Pharmaceutical Products – Appendix on Cell Therapy Products
(Draft for Soliciting Opinions)
Chapter 1: Scope
Article 1 [Scope] This appendix defines cell therapy products (hereinafter referred to as "cell products") as human-derived live-cell products, including cells that have or have not undergone genetic modification—such as autologous or allogeneic immune cells, stem cells, tissue cells, or cell lines. However, this definition excludes blood components used for transfusion, established transplantation-grade hematopoietic stem cells, reproductive-related cells, as well as tissue and organ products composed of cells.
Article 2 [Scope of Application] This appendix applies to the entire process of cell product handling—from the transportation and receipt of donor materials, through product manufacturing and testing, to finished-product release, storage, and shipment.
The processes for the production, testing, and release of gene-modified vectors or other starting biological materials (including viruses, plasmids, RNA, antigen peptides, antigen proteins, protein-RNA complexes, and more) intended directly for cell product manufacturing must comply with the requirements outlined in the current version of the "Pharmaceutical Production Quality Management Regulations," its relevant appendices, and this supplementary appendix.
Article 3 [General Requirements] Since the donor materials for cell products are derived from human sources, their production must also comply with relevant national regulations to prevent the introduction or transmission of infectious disease pathogens.
Chapter 2: Principles
Article 4 [Specificity] Cell-based products have the following unique characteristics:
(1) [Safety of Donor Materials] Donor materials are derived from human sources and may contain pathogens that cause infectious diseases;
(II) [Characteristics of the Manufacturing Process] The quality of donor materials varies significantly depending on factors such as their source, type, and inherent characteristics. As a result, the manufacturing process for the product may need to be adjusted accordingly—within the scope of the product’s approved registration—to account for these variations in donor material quality.
(III) [Characteristics of Production Batch Sizes] Due to limitations in the availability and scope of donor materials, product production batches are typically small. As a result, the production organization model tends to be more flexible, with closer integration between manufacturing processes and clinical needs.
(IV) [Impact of Temperature] Temperature has a more significant impact on the quality of donor materials and products;
(V) [Preventing Contamination and Cross-Contamination] The production process following donor material collection involves live cells, making the product highly susceptible to microbial contamination or cross-contamination—contaminants that are particularly difficult to eliminate.
(6) [Preventing Confusion and Errors] Autologous cell products or products manufactured using allogeneic donor materials that require patient matching—once mistakenly mixed up, leading to a mismatch between donor materials/cells and the patient—could result in life-threatening complications for the patient.
Article 5 [Special Controls] Given the unique characteristics of cell-based products, companies should implement special control measures throughout the entire process—from donor material collection to product manufacturing—these measures should include, at a minimum:
(1) Conduct a risk assessment of the entire process—from the receipt of donor materials to the storage and transportation of finished products—and develop corresponding risk control strategies to ensure the safety, efficacy, and controllable quality of the products.
(II) Establish biosafety management systems and records, equip facilities and devices that ensure biosafety, and implement measures to prevent and control biosafety risks during the product manufacturing process, thereby safeguarding against the introduction and spread of pathogens.
(III) Monitor the temperature and operational time limits of products or the production environment throughout the entire process—from donor material transportation and receipt to product manufacturing, storage, and shipping—ensuring that all operations are completed within the specified temperature ranges and time constraints.
(IV) The entire product manufacturing process should pay particular attention to preventing microbial contamination or cross-contamination, including cross-contamination risks posed by the carrier production process to the product itself, as well as potential cross-contamination risks among different carriers during their respective manufacturing stages.
(5) Throughout the entire process—from donor material collection to patient use—the product must be properly labeled and fully traceable, ensuring clarity and preventing confusion or errors.
Chapter 3: Personnel
Article 6 [Qualifications of Key Personnel] The Head of Production Management, the Head of Quality Management, and the Qualified Person for Quality Control should possess relevant expertise (such as microbiology, biology, immunology, biochemistry, or biopharmaceutical sciences) and be fully capable of fulfilling their responsibilities in production and quality management.
Article 7 [Personnel Safety Protection Training] Personnel involved in cell product manufacturing, quality assurance, quality control, and other related activities—including cleaning and maintenance staff—must undergo training in biosafety protection, with a particular focus on knowledge about preventing the transmission of infectious pathogens via donor-derived materials. All training must comply with national regulations governing biosafety.
Article 8 [Restrictions on Personnel Activities] During production, personnel involved in vector manufacturing must not enter the cell product production area unless effective decontamination measures have been implemented as required. Additionally, individuals who have come into contact with donor materials containing infectious disease pathogens are prohibited from entering any other production areas.
Chapter 4: Factory Buildings, Facilities, and Equipment
Article 9 [Factory Zone Design] Gene-modified viral vectors directly used in cell product manufacturing should be processed in separate, dedicated production areas from the cell products and other vectors or biological materials, each equipped with its own independent air conditioning and purification system.
Article 10 [Requirements for Production Facilities Handling Donor Materials Containing Pathogens of Infectious Diseases] When producing cell products using donor materials containing infectious disease pathogens, the manufacturing operations should be conducted in a dedicated, separate production area equipped with an independent air conditioning and purification system, ensuring that the production area where the product is exposed to the environment maintains negative pressure relative to surrounding areas.
Article 11 [Closed System] Cell product production operations should be carried out using closed systems or equipment; the cleanliness level of the environment where these systems or devices are located can be appropriately reduced.
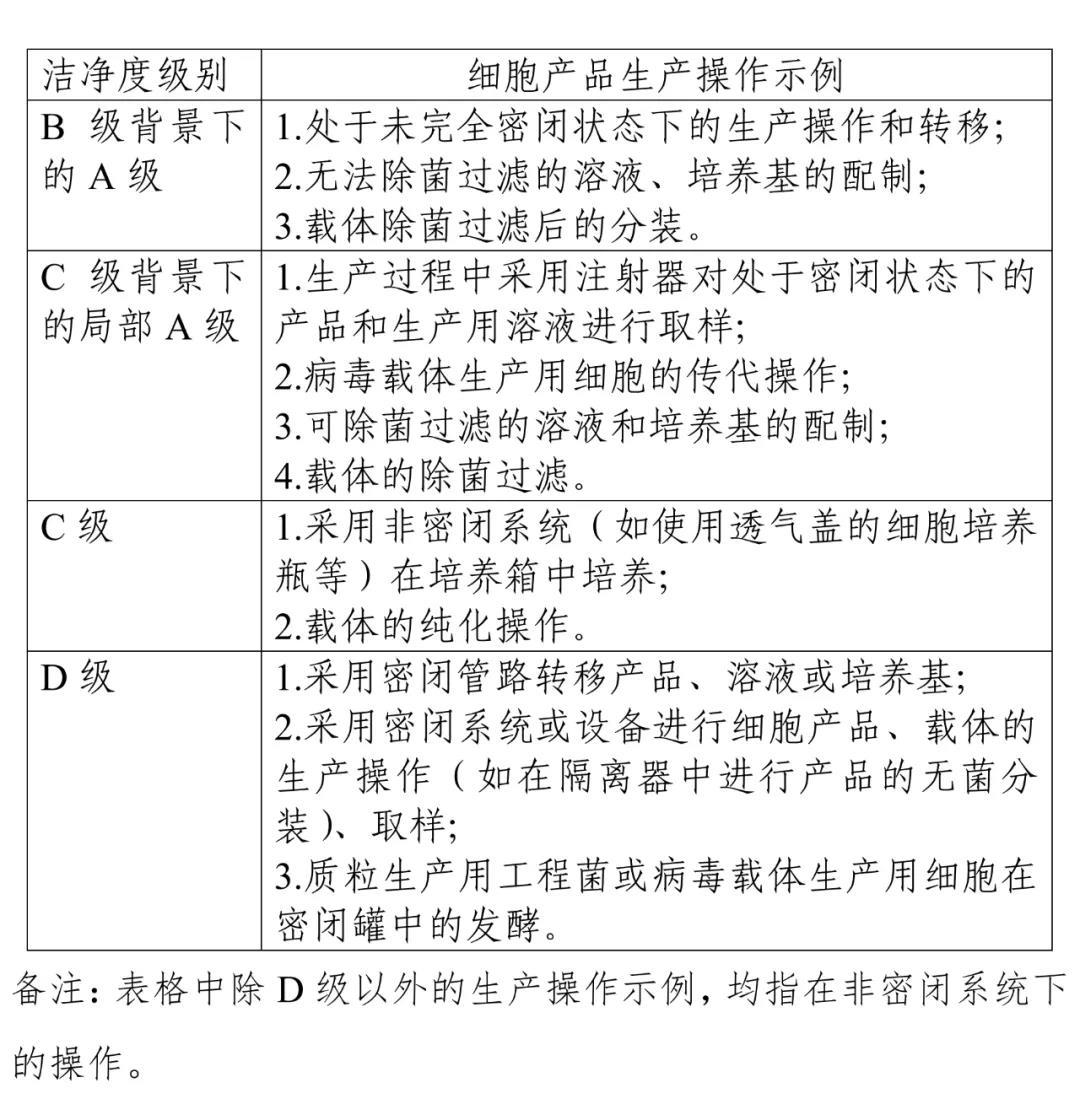
Article 12 [Cleanliness Level of the Production Environment] Cell products, gene-modified vectors directly used in the production of cell products, or other starting biological materials— their manufacturing environment cleanliness levels can be selected based on the examples provided in the table.
Article 14 [Testing Laboratory] The donor screening, donor material, and cell product testing laboratories used for infectious disease pathogen marker assays—or for detecting samples containing infectious agents—must comply with national regulations regarding laboratory biosafety. Where necessary, these facilities should also be equipped with on-site inactivation or disinfection equipment.
Chapter 5: Donor Screening and Donor Materials
Article 15 [Donor Screening Criteria and Donor Material Quality Standards] Enterprises should establish donor screening criteria and quality standards for donor materials in accordance with the requirements for cell product registration approvals. They should also conduct a comprehensive risk assessment, taking into account factors such as the biosafety level of microorganisms, the category of infectious diseases, and the intended use of the cell product, while regularly reviewing the suitability of these measures.
Article 16 [Qualifications of Medical Institutions] Companies should select medical institutions with legitimate qualifications as the facilities for donor material collection and cell product usage, while clearly defining the responsibilities of both parties. The quality management department should conduct a quality assessment of these medical institutions and, in collaboration with relevant internal departments, carry out on-site quality audits to ensure that the institutions’ donor screening processes, donor material collection procedures, and product utilization practices comply with applicable regulations.
Article 17 [Procedure for Accrediting Medical Institutions] Enterprises should establish operational procedures for quality assessment and approval of medical institutions, clearly defining the qualifications required of these institutions, the principles for selection, the methods of quality evaluation, the evaluation criteria, and the process for recognizing qualified medical facilities. Additionally, the procedures should specify the content, frequency, composition, and qualifications of personnel involved in on-site quality audits.
Article 18 [List of Qualified Institutions and Quality Records] The enterprise's quality management department should assign a dedicated person to oversee on-site quality audits of medical institutions, identify a list of accredited and qualified healthcare facilities, and establish a comprehensive quality file for each institution.
Article 19 [Quality Agreement] Companies should sign a quality agreement with accredited, qualified medical institutions. The quality agreement must include at least the responsibilities of both the medical institution and the company, as well as details on donor material collection methods, storage conditions, quality standards, acceptance procedures, and product usage guidelines.
Article 20 [Revocation of Medical Institution Qualification] Enterprises should regularly review and assess how medical institutions collect donor materials and use their products. If any non-compliance with operational procedures is identified—particularly when such issues could potentially harm patients—companies must promptly request the medical institutions to implement corrective and preventive measures. If necessary, these institutions may no longer be included on the list of qualified medical facilities.
Article 21 [Collection Operating Procedures] Companies should establish written guidelines for donor material collection, transportation, and receipt, detailing the methods for collecting donor materials, as well as the specific storage and transportation conditions and acceptance criteria.
Article 22 [Recipient of Donor Materials] Enterprises should inspect at least the following items for each batch of donor materials received:
(1) Sourced from legally recognized medical institutions that have been evaluated and approved by the company;
(II) Temperature and time-limit monitoring records during transportation are complete, ensuring that temperature and time limits comply with regulatory requirements. Additionally, if there are specific storage temperature requirements for donor materials immediately after collection, comprehensive temperature-monitoring records must also be maintained—and these must meet the applicable standards.
(III) Packaging is intact and undamaged;
(IV) The packaging label must include complete information, including at least an individual identification code traceable back to the donor, the date and time of collection, the volume collected, and the name of the medical institution where the collection was performed. If a computerized system is used, the packaging label must also enable traceability to the aforementioned details.
(5) Donor Material Collection Records;
(6) Clinical test results for donor screening must include at least the findings from tests specific to infectious disease pathogens.
Article 23: [Materials from Positive Donors] 】 Companies must isolate and store autologous donor materials known to contain infectious disease pathogens, with each package clearly labeled.
Enterprises must not accept allogeneic donor materials that do not meet registration standards.
Article 24 [Quality Assessment] Before putting into production and use, companies should conduct a quality assessment of each batch of donor materials, which must include at least the following:
(1) Verify that the donor materials originate from legally operating medical institutions approved by the company and that the donors meet the screening criteria; also, cross-check the relevant information as outlined in Article 21, Paragraph (4).
(II) Temperature monitoring records during transportation are complete and meet the specified requirements. If there are special temperature storage requirements for donor materials immediately after collection, complete temperature monitoring records must also be maintained, ensuring compliance with relevant standards.
(III) The storage temperature and duration of donor materials, from the time they are collected at the medical facility until release by the company for pre-production use, must comply with the specified requirements.
(IV) Donor materials are packaged intact, with no damage;
(5) Deviations occurring during transportation and storage have been investigated and addressed according to the relevant procedures.
Article 25: [Isolation of Positive Donor Materials] 】 Donor materials containing infectious disease pathogens should be kept isolated from other donor materials during transportation, receipt, storage, distribution, or shipment.
Chapter 6: Materials and Products
Article 26 [Raw Material Control] Biological materials used in cell product manufacturing—such as cell lines, genetically engineered bacteria, vectors, and animal-derived reagents and sera—must be sourced legally and safely, while also meeting quality standards to prevent the introduction or spread of infectious disease pathogens.
Article 27 [Critical Materials] Companies should conduct risk assessments for materials to identify critical inputs—such as gene-modified vectors or other starting biological materials directly used in cell product manufacturing, as well as cytokines, growth factors, enzymes, serums, and more. The identification of these critical materials must be documented. Incoming inspections should be performed on all critical materials, with specific risks and additional risk-mitigation measures (e.g., enhanced quality control) taken into account.
Article 28 [In Vitro Diagnostic Reagents] In vitro diagnostic reagents used for the detection of specific infectious disease pathogens (HIV, HBV, HCV, and Treponema pallidum), along with their associated management guidelines, must utilize in vitro diagnostic reagents approved by the national drug regulatory authority.
Article 29 [Transportation Confirmation] The transportation of donor materials and cell products must be properly verified.
Article 30 [Handling of Nonconforming Materials] Safety and efficient operating procedures should be established for handling non-compliant donor materials, intermediate products, and finished goods, with all handling activities properly documented.
Chapter 7: Production Management
Article 31 [Division of Batches] Cell products can be defined as a batch—based on their process characteristics—when they consist of a uniform-quality product produced in a single production cycle, using the same manufacturing process and under identical production conditions. The total number of cells produced within a single batch constitutes the batch size for that particular production run.
Article 32 [Process Adjustment] When adjustments to the manufacturing process parameters of cell products are required due to inherent quality variations in donor materials, companies must make these adjustments within the scope approved during product registration.
Article 33 [Sterile Process Simulation] Sterile process simulation studies for cell products, gene-modified vectors directly used in the production of cell products, or other starting biological materials should meet at least the following requirements:
(1) For sterile production operations conducted in non-enclosed systems, the aseptic process simulation test should include all exposed steps involving manual handling.
(2) For processes conducted using a closed system for sterile production operations, the sterility process simulation study should focus primarily on steps involving connections to the closed system. If any sterile production operations remain un-simulated, a risk assessment must be performed, and the rationale for not simulating these steps must be documented in writing.
(III) For sterile production operations that require an extended duration, a risk assessment should be conducted to justify shortening the duration of certain simulated processes, such as centrifugation or cultivation.
(IV) Sterile production operations (such as cryopreservation) that inhibit microbial growth and could potentially affect the results of sterile process simulation tests may, following a risk assessment, be excluded from the media simulation tests.
(V) When multiple identical production lines are located in the same production area, each line can conduct a sterile process simulation test again every six months per shift—either using the extreme-value method, the matrix method, or a combination of both—after successfully passing the initial validation of the sterile process simulation test.
Different products manufactured using the same equipment and process steps can undergo sterility process simulation testing using the extreme-value method, which replicates the most challenging conditions of specific production operations. Alternatively, when employing the matrix method for sterility process simulation, the worst-case conditions for similar process steps should be simulated. If both methods are used in combination, the rationale and its justification must be documented in writing. Importantly, the simulation must cover all sterile manufacturing operations under the most critical conditions, as well as include every type of equipment used in production.
Article 34 [Process Validation] Cell product manufacturing processes should be validated, and this process validation must meet at least the following requirements:
(1) The manufacturing process for cell products using autologous donor materials has certain unique characteristics; therefore, the donor materials used for validation can be sourced from healthy volunteers. If the materials are derived from patients, a simultaneous validation approach may be employed.
(II) The worst-case conditions in actual production should be considered based on a risk assessment. For instance, if multiple identical production lines are located in the same production area, or if several isolators or closed systems are installed within the same production room, the maximum number of production lines—or the number of isolators or closed systems—that can operate simultaneously must be determined. Additionally, factors such as the production environment, personnel capabilities, and laboratory testing capacity should also be evaluated as part of these worst-case scenarios and thoroughly validated.
Article 35 [Carrier Process Validation] Gene-modified vectors or other starting biological materials directly used in cell product manufacturing must undergo validation, with the process validation including at least three consecutive batches produced under a complete manufacturing process.
Article 36 [Prevention and Control of Contamination and Cross-Contamination During Production] Measures should be implemented during the cell product manufacturing process to prevent contamination and cross-contamination as much as possible, while also controlling quality risks, such as:
(1) Autologous donor materials containing infectious disease pathogens must not come into contact with other donor materials or cell products that do not contain infectious disease pathogens during production and transportation.
(II) When production is carried out using non-enclosed systems or equipment, different cell products must not be manufactured simultaneously within the same production area; similarly, different batches of the same cell product cannot be produced concurrently in the same production room.
(III) When multiple isolators are located within the same production area, their integrity should be regularly checked. Isolators must not exhaust air directly into the operational room, and any exhaust air should not be recycled. Additionally, effective measures must be implemented to prevent errors and cross-contamination involving materials, products, and waste—for example, by using sealed transfer systems, alternating operations, designated storage areas, sterilization and disinfection protocols, and unidirectional airflow transfer methods.
(IV) Multiple biosafety cabinets located within the same production area but housed in separate operational rooms should ideally utilize a closed system to simultaneously produce different batches of the same cell-based product. If full containment control throughout the entire production process cannot be ensured, a thorough risk assessment must be conducted, accompanied by robust measures designed to prevent errors and cross-contamination of materials, products, and waste—such as sealed transfer systems, precise room pressure differential management, strict no-cross-room-operation protocols, avoidance of overlapping personnel movement, rigorous sterilization and disinfection procedures, and implementation of unidirectional airflow for material transfer.
(5) When using a closed system within the same production area for manufacturing different batches of the same cell product, multiple identical or even different production steps should be avoided simultaneously in the same operational room—except for the cell culture step itself. After completing each production step, thorough cleaning and sanitization must be performed promptly. Additionally, effective measures should be implemented to prevent errors and cross-contamination involving materials, products, and waste, such as maintaining appropriate room pressure differentials, controlling personnel access, staggering operations, enforcing fixed-location management, conducting regular sterilization and disinfection procedures, and utilizing unidirectional airflow systems for material transfer.
(6) When using a closed system for cell culture, different batches of products can be cultured and stored simultaneously in the same production area or incubator—provided that effective measures are in place to prevent any potential mix-ups. In contrast, if an open (non-closed) system is used for cell culture, physical separation must be implemented between different batches within the incubator (e.g., by using a honeycomb-style incubator) or dedicated incubators should be assigned to individual production batches. Additionally, the incubator environment must maintain a high level of cleanliness and be suitable for disinfection or sterilization procedures. A thorough risk assessment should also be conducted to ensure that cross-contamination and confusion are effectively avoided.
(7) In the event of accidental opening or leakage in a closed system or equipment, a risk assessment must be conducted, followed by the implementation of effective emergency measures.
Article 37 [Handling of Microbial Contamination] Monitoring procedures for microbial contamination in each production process should be established, along with clearly defined corrective actions. These procedures must specify the conditions under which normal production can resume after contamination has been eliminated. When handling contaminated products or materials, any extraneous microorganisms detected during the production process should be identified, and their potential impact on product quality must be thoroughly assessed.
All records of microbial contamination and handling during production should be maintained.
Article 38 [Prevention and Control of Confusion and Errors in Production] Measures should be implemented during the cell product manufacturing process to prevent confusion and errors as much as possible, such as:
(1) Donor materials and products used during the manufacturing process should all be properly labeled, and products stored at low temperatures must also bear identification tags.
(II) The donor materials and products must include a unique, identifiable number (or code) that links back to the donor;
(III) Before production, carefully verify the donor materials and product identification information, especially the unique identifier (or code) used to recognize the donor. Verification must be documented.
(IV) During the production process, if products need to be labeled, the accuracy of the information on the label must be verified to ensure it matches the unique identifier (or code) used to identify the donor on the donor material. Verification must be documented.
Article 39 [Timely Visual Inspection] Packaging containers used for cell product manufacturing, along with any connected containers (if applicable), should undergo immediate visual inspections both before use and after filling to check for signs of damage or contamination. Visual inspection results must be documented.
Article 40 [Single-Use Consumables] Sterile consumables that come into direct contact with cell products should use single-use materials whenever possible.
Article 41 [Transshipment of Intermediate Goods] For intermediate products and materials in the production process that have specific handling requirements—such as temperature or time constraints—clear regulations must be established for the transportation conditions. Additionally, appropriate monitoring or control measures should be implemented during transit, accompanied by thorough documentation.
Article 42 [Waste Disposal] Contaminated materials, waste, or suspiciously infected items containing infectious pathogens during the production process must be disinfected on-site and fully inactivated before being removed from the work area. The handling process must comply with national regulations governing the management of hazardous waste.
Chapter 8: Quality Management
Article 43 [Sample Retention] Donor materials for cell products, critical raw materials, and finished products should be retained according to regulations. In special cases—such as when donor materials or raw materials are scarce, product batches are small, shelf life is short, or clinical needs urgently require it—the volume of retained samples, sample packaging, storage conditions, and retention period for donor materials, raw materials, and cell products may be appropriately adjusted as follows:
(1) Retained samples of donor materials
Donor materials from both autologous and allogeneic sources should generally be retained as samples. For rare donor materials, if there is a need to adjust the sampling strategy or decide against retaining samples altogether, a written explanation outlining the rationale must be provided.
(II) Sample Retention of Materials
Critical materials—such as gene-modified vectors or other starting biological materials directly used in cell product manufacturing, as well as cytokines, growth factors, enzymes, serums, and more—are essential for identifying potential quality issues with the investigational product. Therefore, companies should retain sample batches within their expiration or shelf life.
(III) Sample Retention of Finished Products
1. The amount of finished product retained for sampling can be appropriately reduced;
2. If it is genuinely impossible to retain a sample due to clinical necessity, a photo of the finished product should be included in the batch record, clearly showing all the complete information from the product label.
3. If it is necessary to shorten the retention period for sample storage, the company must conduct an evaluation and provide a corresponding report.
4. When a product has a short shelf life and its retained sample needs to be stored for an extended period, appropriate methods (such as low-temperature cryopreservation) should be employed to ensure the sample continues to serve its intended purpose. For instance, freshly collected cells may lose their suitability as quality-characterization samples after cryopreservation, but they can still be used effectively for sterility testing or viral detection. If retaining finished products in frozen form no longer meets the original objective, companies should explore alternative approaches—such as using intermediate products or differentiated cells—as substitutes for the original finished-product samples.
5. If finished-product samples cannot be used, intermediate products with the same composition as the finished product may be selected for retention. The packaging, storage conditions, and shelf life of these retained samples must meet the intended purpose and requirements. Additionally, the packaging method and materials should be identical to or closely resemble those of the marketed product.
Article 44 [Pre-Release Quality Assessment of Products] Cell products should undergo testing according to the requirements of the registration standards. Before release, the quality assessment must verify that all information for each batch is complete, accurate, and traceable; otherwise, the product must not be released.
Cell products derived from the patient’s own cells, or those produced using allogeneic donor materials intended for patient-specific use, must have their donor material or cell source information verified by the company prior to release—and their compatibility with the patient must be confirmed.
Quality evaluations conducted before release for cell products manufactured using donor materials put into use prior to completion of testing should assess the impact of these donor materials on the final product quality.
Article 45 [Record Keeping] Batch records for cell products should be retained for at least five years beyond the product's expiration date.
Cell products manufactured using allogeneic donor materials must have their batch records archived for long-term preservation.
Article 46 [Handling of Quality Defects] Enterprises should establish emergency response procedures. When it is discovered during the transportation or use of cell products that there are quality defects—such as damaged packaging bags, incorrect or missing label information, or temperature deviations exceeding specified limits during transit—they must immediately activate the emergency response and initiate a thorough investigation. All relevant emergency actions and investigations must be documented and reported. If necessary, a product recall should also be launched.
Chapter 9: Product Traceability System
Article 47 [Product Traceability System] Enterprises should establish product identification and traceability systems to ensure that products from different donors are not mixed up or misidentified throughout the entire process—from donor material transportation and receipt to product manufacturing and use. This will also guarantee the proper matching between donor materials/cells and patients, while maintaining full traceability at every stage.
The system should utilize a validated computerized system capable of enabling two-way traceability of products—from donor to patient or from patient to donor. This includes the entire process, from receiving donor materials, through transportation, manufacturing, testing, and release, all the way to the final product’s shipment and use.
Article 48 [Unique Donor Number] Companies should assign a unique number (or code) to each donor, used to identify donor materials and products.
Article 49 [Written Operating Procedures] Companies should establish written operating procedures that outline the proper labeling and verification of donor materials and products—covering processes such as receipt, transportation, production, testing, release, storage, and distribution—as well as the associated documentation. These procedures must ensure that uniquely identifiable donor numbers (or codes) are correctly applied without errors or omissions, guaranteeing accurate matching between donor materials/cells and patients while maintaining full traceability throughout the entire process.
Article 50 [Information Exchange] Enterprises should establish an information-sharing mechanism with medical institutions to promptly exchange key details such as donor material collection, product usage, and critical information related to product quality, taking appropriate measures when necessary.
Chapter 10 Other
Article 51 [Product Usage Guidance] Companies should develop detailed product usage instruction manuals. For products that require on-site preparation before use in medical facilities, the operating procedures must be described thoroughly—such as cell recovery, dilution, and washing methods—as well as the preparation environment, aseptic handling requirements, recommended storage temperature and duration, and transportation methods. Where necessary, visual aids like images or videos may also be included for clarification.
Article 52 [Training] Enterprises should provide training and conduct assessments for medical institution personnel on donor material collection requirements and product usage, and records of these training sessions and assessments must be maintained.
Chapter 11: Technical Terms
Article 53 [Definitions] The meanings of the following terms are:
(1) Donor
Refers to individuals who provide cells or tissues used in the production of cell-based products, and these individuals can be either healthy or patients.
(II) Donor Materials
Refers to cells or tissues—among other materials—obtained from donors who meet the screening criteria, intended for use in cell product manufacturing.
(III) Autologous Cell Products
Refers to cell products that are produced and processed from cells collected from the patient, then reintroduced back into the patient's body.
(IV) Production Area
Refers to a specific set of rooms within a building designated for production operations, typically equipped with an air-conditioning system—including ventilation, temperature control, and essential humidity regulation—as well as necessary air filtration and purification.
(V) Closed System
A system designed and operated to prevent products or materials from being exposed to the indoor environment. When products or materials are transferred into this enclosed system, it must be done in a non-exposure manner—such as via sterile connectors or a sealed transfer system—to ensure they remain protected from contact with the surrounding indoor air. If the sealed system needs to be opened—for tasks like installing filters or making connections—it must be thoroughly disinfected or sterilized before being resealed or reused.
Reprint Notice: This article is reprinted from the official website of the National Medical Products Administration. If there is any infringement, please contact us for removal.
Previous:
Related News

Here is the title—h1 placeholder text


Copyright © Jiuzhitang Maker (Beijing) Cell Technology Co., Ltd.
Powered by: 300.cn SEO | Privacy Policy